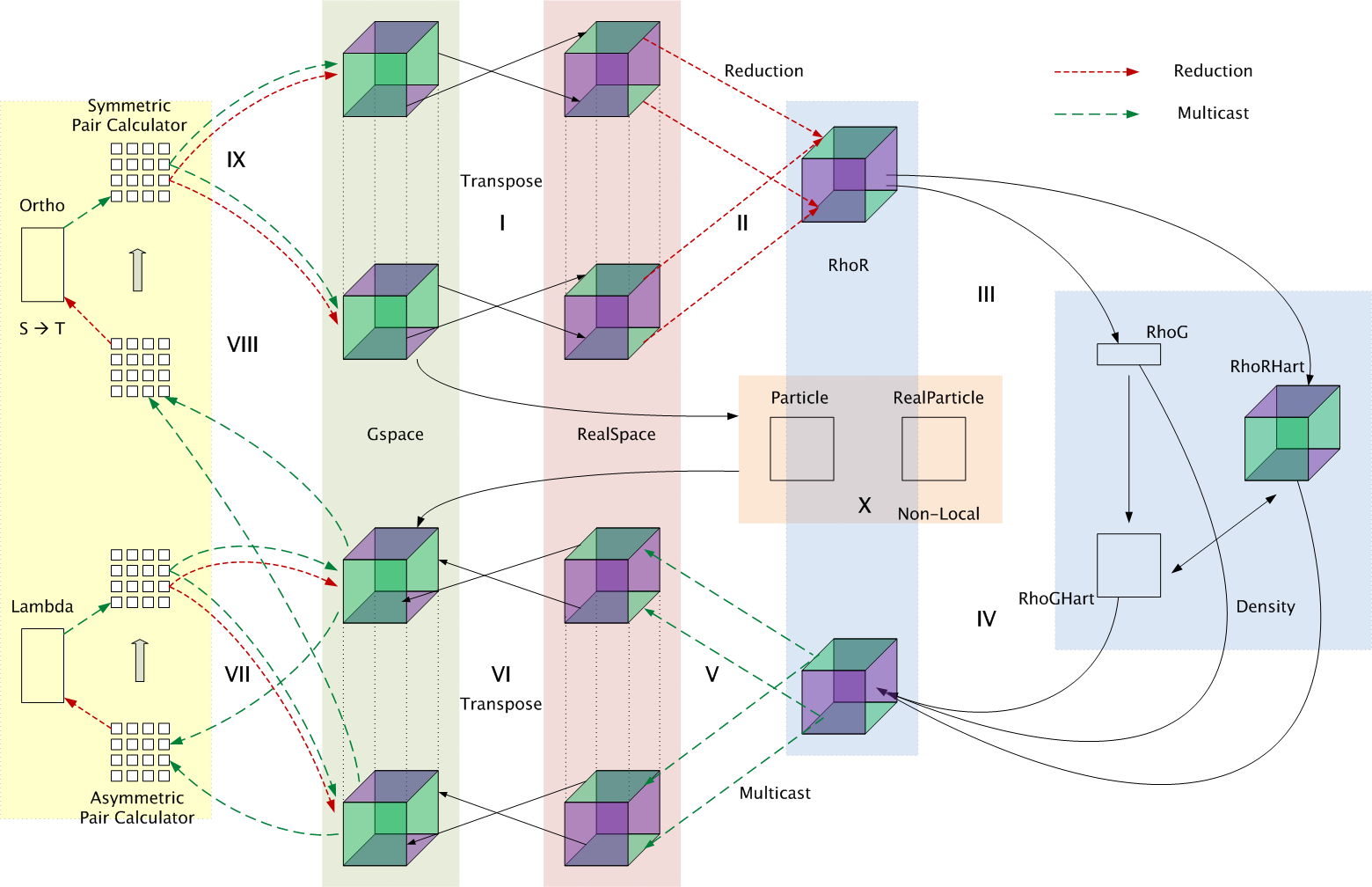
The message-driven object-based overdecomposition approach embodied in the
Charm++ parallel programming system forms the backbone of OpenAtom. Work is
divided into a large number of objects and the computation is expressed in
terms of these objects only. These objects, termed chares, are organized
into indexed collections, called chare-arrays. In this way, Charm++ separates
the issue of decomposition and where the decomposed objects are placed.
Either the runtime system or the user specifies where the objects are placed.
As shown in the Figure on the right, in phase I, the electronic states are
transformed from their g-space representation to their real-space state
representation. The parallel 3D-FFTs employed to effect this change of
representation are implemented using a transpose-based method. Since the
states have a sparse spherical distribution within their 3D g-space grid but
are dense in real space, this order of operations reduces the communication
cost. The reduction to form the probability density in real space occurs in
phase II. Both the g-space and the real-space representations are defined by
different chare-arrays that are suitably mapped onto real processors to
optimize communication. Once the states are available in real-space, the
electronic density can be computed via a simple aggregation across all the
states.
Decomposition in OpenAtom: physical entities
encapsulated as chares are defined; the control flow is described among
the chares.
In phases II-VI, the electron density is processed. The electron density in
real space is immediately used to compute a portion of the
exchange-correlation energy upon arrival. The Fourier components of the
density are then created by subjecting a copy of the density in real space to
a 3D-FFT (phase IV). Once in gspace, the Fourier coefficients of the density
are used to compute the Hartree and external energies via more 3D-FFTs, which
can be performed independently. In addition, the gradient of the electron
density in real space is created by making three copies of the Fourier
coefficients of the density, point by point multiplying each appropriately
and performing three 3D-FFTs to real space. The electron density and its
gradients are employed to compute a second, gradient-dependent contribution
to the exchange-correlation energy. The Kohn-Sham potential is created by
performing 5 more 3D-FFTs (requiring 10 transposes) on data sets generated
during the energy computation. Each of these computations is performed by
chare-arrays that are mapped to different processors, so that concurrent
computations can overlap with each other and reduce communication overheads.
After performing multiple 3D-FFTs (phase VI) on the product of the Kohn-Sham
potential with each of the states, in a procedure exactly the reverse of that
used to form the density, the forces are obtained in g-space.
Phase IX requires the computation of the kinetic energy of the
non-interacting electrons which can be computed without interprocessor
communication and the computation of the non-local interaction between the
electrons and the atoms via the new EES method. The latter calculation
involves many more 3D-FFTs and is interleaved with Phases II-VI. The force
regularization and orthonormalization are performed in phases VII-VIII. Once
the forces have been computed, a series of matrix multiplications must be
performed. For functional minimization, the forces are multiplied by the
states, result of which is used to modify the forces. Under non-orthogonal
state dynamics the forces are further multiplied by the precomputed inverse
square root matrix which is available due to the change in the order of
operations required by the dynamics method as described below. The modified
forces are then employed to evolve the states in both cases. Under
functional optimization, the evolved states are slightly non-orthogonal and
must be reorthogonalized by matrix transformation. This requires the
computation of the overlap matrix, S. Next, the inverse square root of the
S-matrix, T is computed and the orthonormal states, are constructed.
Decomposition: OpenAtom's nine phases are parallelized by decomposing
the system into 14 indexed collections of objects, each implemented as a
chare array. The real-space and g-space representations of the electronic
states are decomposed into the 2D chare arrays where the first dimension is
the number of states, and the second dimension is used to divide each state
among smaller sets. The real-space and g-space representations required for
the non-local work are decomposed as the states. The g-space of electronic
density is divided among 1D chare array in order to load balance the g-space
points without the cutoff. The real-space in contrast is dense, and is
divided among a 2D chare array in order to allow fine-grained parallelism.
The real-space and g-space representations required for the external/Hartree
energy work are decomposed as the density but with an additional atom type
index. Finally, the orthogonality related computation is decomposed into two
sparse 4D g-space based arrays and a 2D auxiliary chare array.
Several scientific methods, Parallel Tempering sampling to treat rough energy
landscapes, Path Integrals to treat nuclear quantum effects, Spin density
functional to treat systems with unpaired electrons, and k-point sampling of
the BZ to treat metals and/or small systems, share a commonality in that
they each consist of a set of slightly different, but mostly independent,
instances of the standard AIMD computation. These instances are
synchronized in different ways for each method, but those differences
comprise relatively small differences in the flow of control.
To support these different multi-instance methods, we have implemented an
overarching (Uber) indexing infrastructure which allows these instances to
reuse all the objects that implement AIMD in OpenAtom while maintaining
distinct sets of instances of the chare arrays comprising each simulation
instance. Furthermore, these are composable, so that more than one type of
multi-instance method can be used in any given simulation, e.g. spin,
k-points, parallel tempering and path integrals functionality might be used
to a science problem such as catalytic activity at a metal surface involving
hydrogen adsorption or abstraction.
Recently, we have developed novel generalized schemes that improve upon the
old topology-aware mapping schemes in OpenAtom in two ways: portability to a
large set of current supercomputers and lesser time to compute the placement.
This has helped improve OpenAtom’s performance on current generations of
systems that have higher dimensionality, e.g. Blue Gene/Q has 5D-torus, and
irregular allocations, e.g. on Cray XE6/XK7 systems, typical allocations are
not restricted to an isolated high bisection bandwidth cuboid.
The major principal underlying the new generalized optimization scheme is the
separation of the logic for mapping from any assumption regarding the
underlying interconnect topology. For example, the mapping code takes several
decisions based on relative closeness of a pair of processes. How the
closeness is defined and computed is left to the topology manager. This
separation allowed the definition of generic rules of thumb on relative
placements of objects of various types (e.g. state, density, pair calculators
or overlap computations) with respect to other objects of the same type and
objects of other types as described below.
When multiple instances are executed concurrently, we have experimented with
multiple schemes to divide the given set of cores among the instances. We
found that the communication among instances is infrequent and often
insignificant, hence the optimal division of cores among instances is more
dependent on the best way individual instances can be placed. Hence, the new
mapping scheme attempts to divide the given allocation into smaller
locality-aware subdomains, wherein the topology-aware mapping is performed
independently.
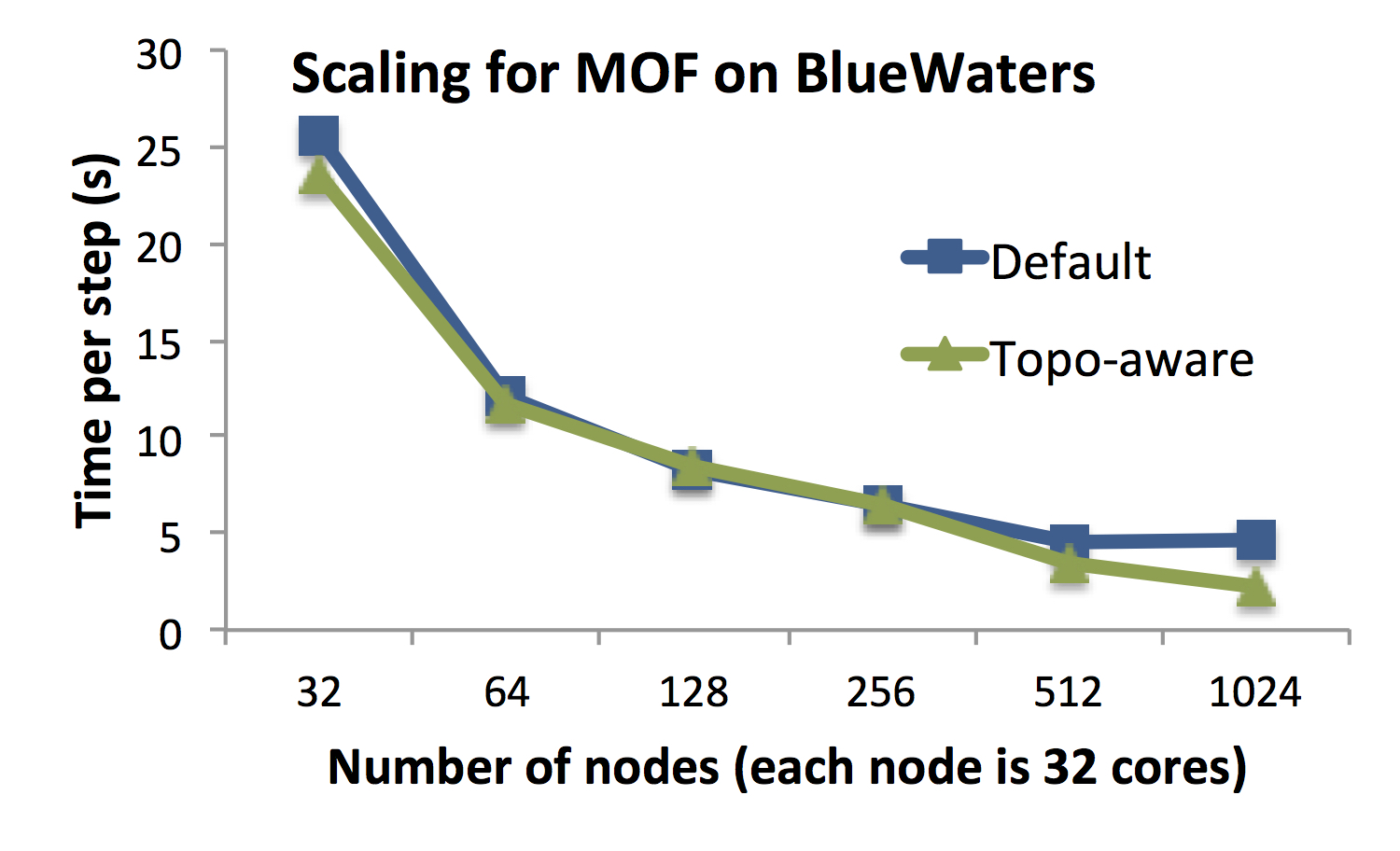
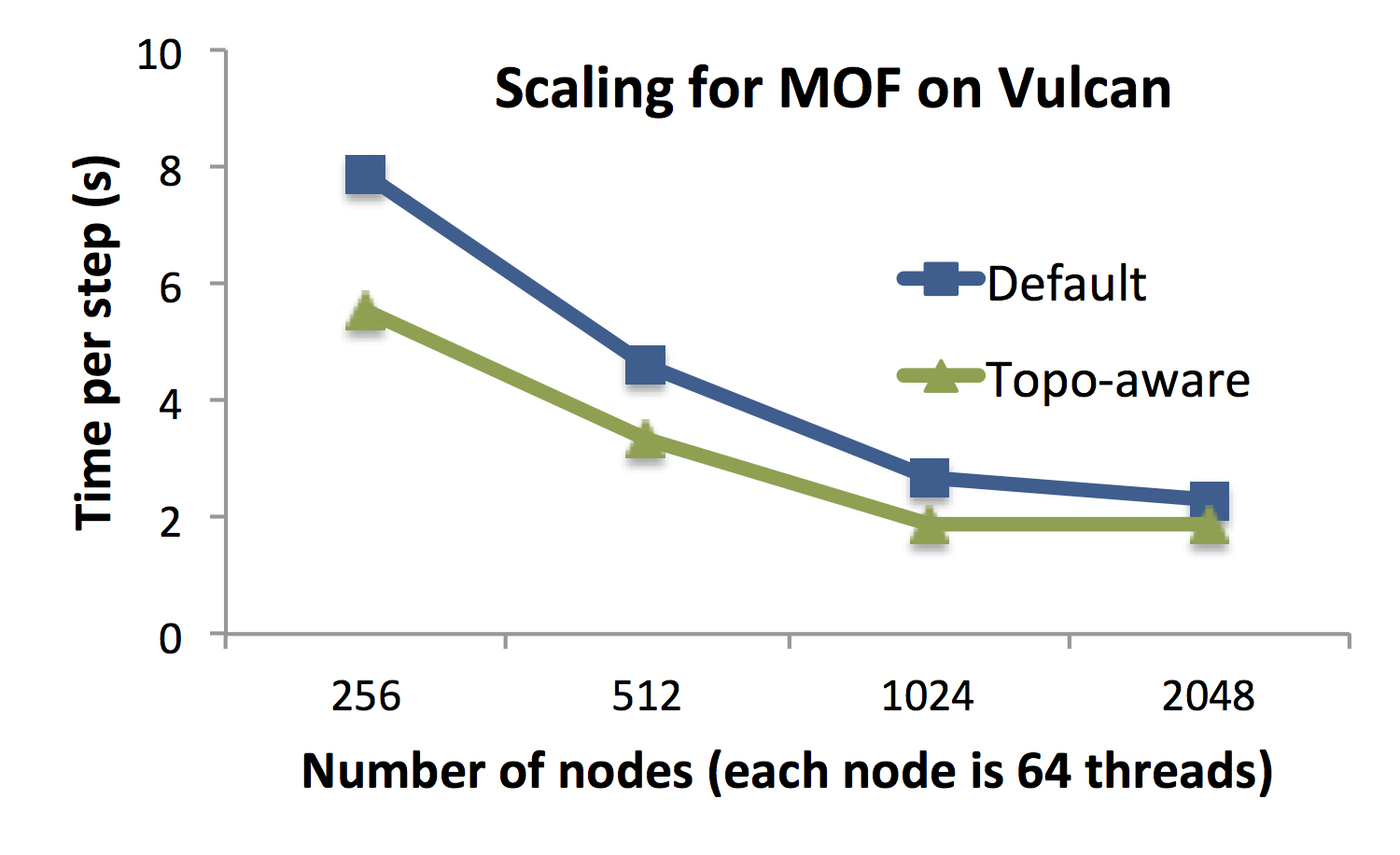
Figure A shows the scaling of OpenAtom on Blue Waters (Cray XE6/XK7 at
NCSA) and Vulcan (Blue Gene/Q at LLNL) for simulating an example MOF system
with 424 atoms and 85 Ry cutoff. On both systems, we observe a significant
reduction in time per step as we scale from lower node counts to 1024
nodes. The time per step reduces by 65% as we scale from 256 nodes of Blue
Gene/Q to 1024 nodes. Similarly, on Blue Waters, the time per step is
reduced to 2.1 seconds from 23.6 seconds as OpenAtom is scaled from 32
nodes to 1024 nodes.
Figure A: For MOF system with 424 atoms, OpenAtom shows good scaling on both
Blue Gene/Q and Cray XE6. When the bisection bandwidth does not increase
with the node count (e.g. on Vulcan from 1024 nodes to 2048 nodes),
the performance does not improve.
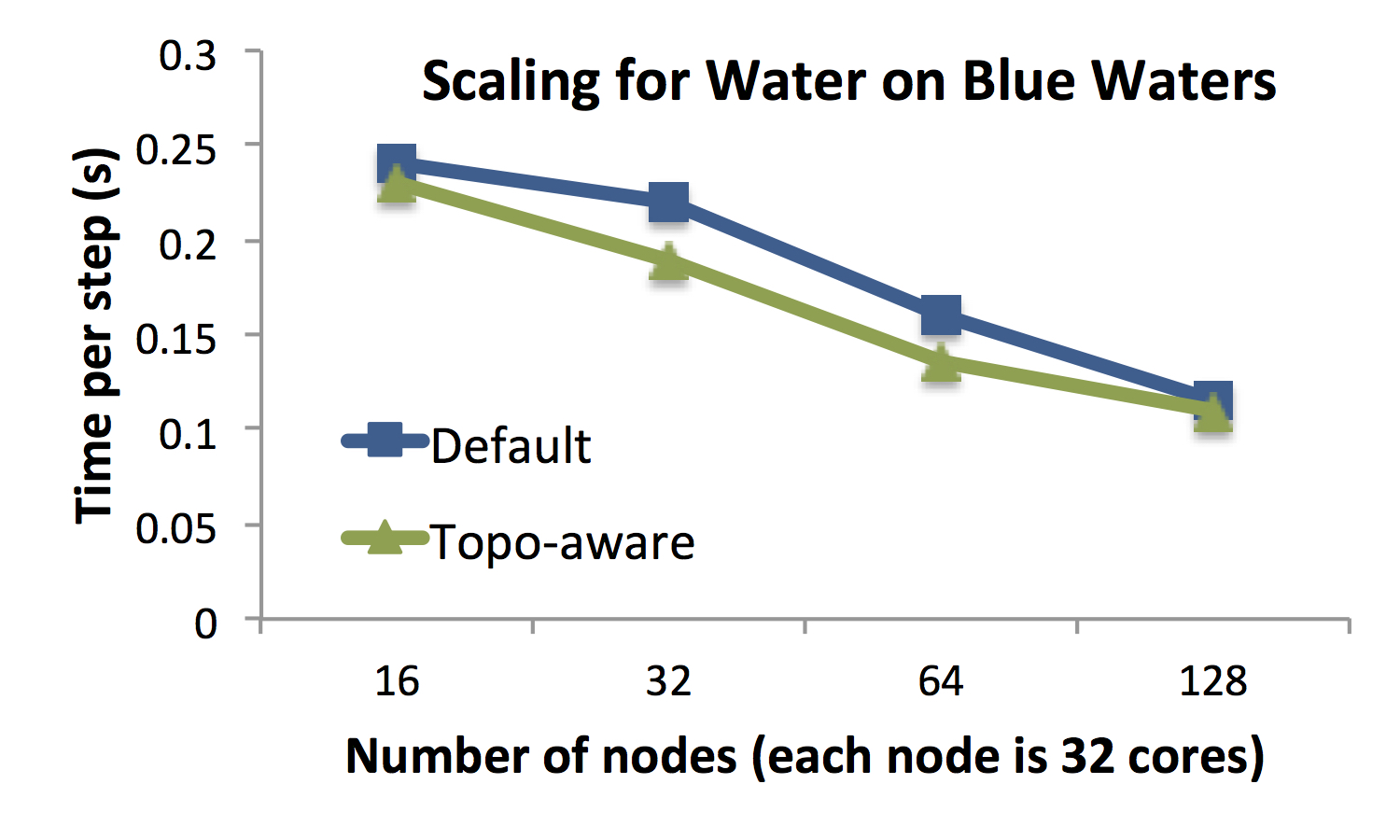
Figure B illustrates OpenAtom’s strong scaling abilities by plotting the
walltime per time step for simulating only 32 molecules of water with 70
Rydberg cutoff radius on Blue Waters. The strong scaling performance of
Path Integral Beads, as well as the impact of topology aware mapping, is
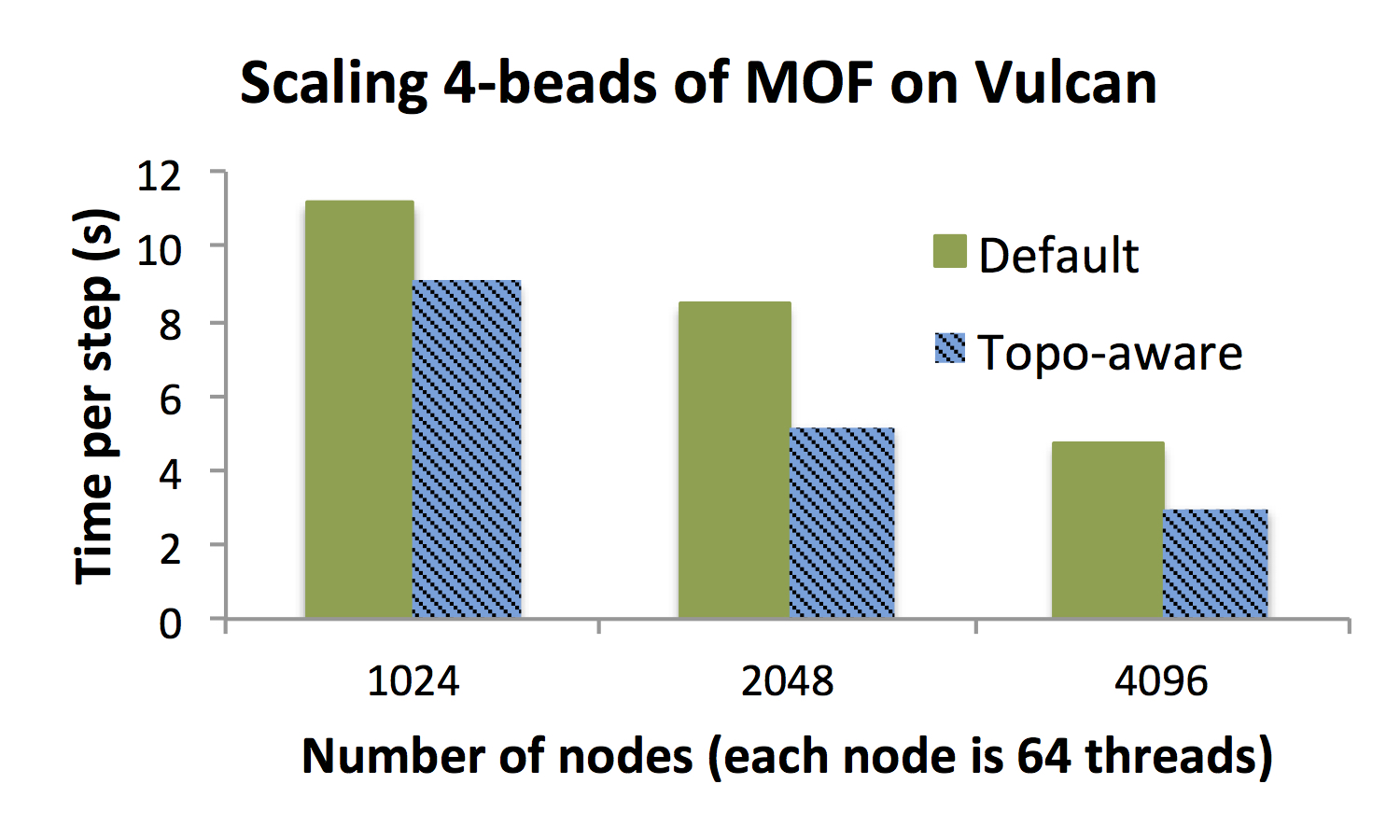
shown in Figure C. A four bead system was chosen for this experiment
in order to fit within the memory constraints of the Blue Gene/Q
architecture. In typical usage, many more beads may be desirable. The
amount of performance improvement gained (18%, 38% and 40%, respectively)
from the use of the topology aware optimization increases with the number
of nodes. This is due to the fact that strong scaling performance for this
communication intensive application is bounded by the network, and this
optimization has the net effect of using the network.
Figure B: Good strong scaling of a small Water system simulation with
32 molecules and a 70 Ryd plane wave cutoff.
Figure C: Scaling results for executing a 4 bead path integral
simulation of MOF.
General purpose codes for performing parallel 3D-FFTs in HPC community (and in
OpenAtom) are based on 1D-decomposition of data. Hence, the amount of
parallelism available for state FFTs is O(nstates * numPlanes). However, the
maximum parallelism available for density is only O(numPlanes) since all
states are combined to form density. This limits the scalability of the
density-related phases of OpenAtom significantly, especially for
large machines with powerful compute nodes.
In order to eliminate the above mentioned scaling bottleneck, we have
developed a fully asynchronous Charm++ based FFT library, Charm-FFT. This
library allows users to create multiple instances of itself and perform
concurrent FFTs with them. Each of the FFT calls runs in the background as
other parts of user code execute, and a callback is invoked when the FFT is
complete. The key features of this library are:
2D-decomposition to enable more parallelism: at higher core
counts, users can define fine-grained decomposition to increase the amount of
available parallelism and better utilize the network bandwidth.
Cutoff-based reduced communication: the data on which FFT is being
performed generally has a science-based cutoff in reciprocal or g-space,
e.g. density has a g-space spherical cutoff, and the library can improve
performance by avoiding communication of data beyond the cutoff points.
User-defined mapping of library objects: the placement of objects
that constitute the FFT instance can be defined by user based on the
application's other concurrent communication.
Overlap with other computational work: given the callback-based
interface and Charm++'s asynchrony, the FFTs are performed in the background
while other application work can be performed.
#Objects
Decomposition
Time (ms)
100
10 x 10
80
300
300 x 1
76
300
75 x 4
69
300
20 x 15
45
400
20 x 20
35
900
30 x 30
24
1600
40 x 40
22
2500
50 x 50
22
3600
60 x 60
23
Effect of decomposition on time to perform FFT on a
300 x 300 x 300 grid. Representative best values are shown for
each of the object counts.
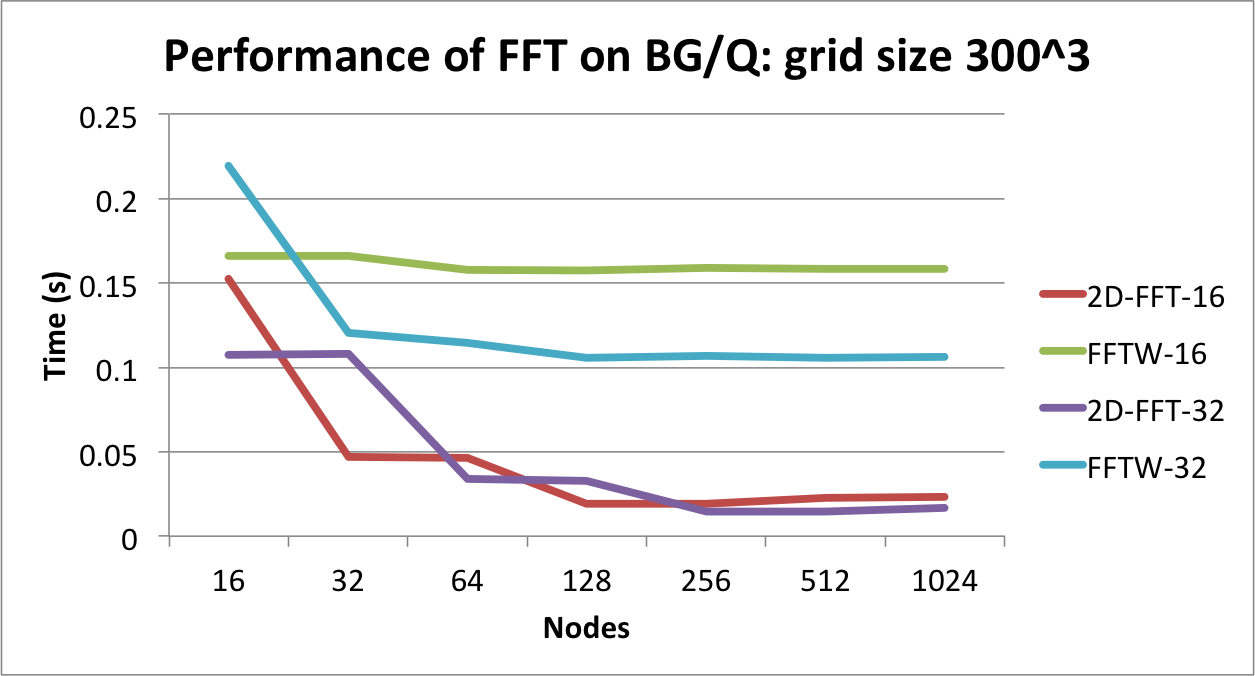
Comparing the performance of parallel FFTW with
Charm++'s pencil-based parallel FFT on IBM BlueGene/Q.
In the table above, we show that by decomposing the 3D grid along two
dimensions, the time to FFT reduces significantly. The best performance
is obtained when we divide the grid among 2,500 objects that are arranged
as a 2D grid of size 50 x 50. An important observation from the table
is the performance difference between two cases in which the same
object count is used, but with different layouts. A 20 x 15 decomposition
improves the FFT performance by 40% over a 300 x 1 decomposition.